


Nuevo Tratamiento: Moduladores
La Fibrosis Quística (FQ) es una enfermedad genética de herencia autosómica recesiva. Es producida por mutaciones en el gen CFTR
localizado en el cromosoma 7. Este gen contiene la información para sintetizar una proteína denominada también CFTR que actúa como un canal localizado en la
membrana apical de las células epiteliales en diferentes órganos (glándula de sudor, bronquios, páncreas, etc.)
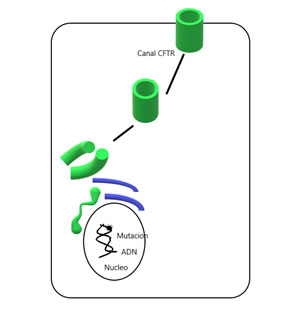
Esta proteína comienza su síntesis en el núcleo, madura en el retículo endoplásmico y aparato de Golgi y se dirige a la membrana para permitir
el paso del cloro mediante la apertura del canal durante el tiempo adecuado cuando es activada por el AMPc.
Las alteraciones en el ADN (mutaciones) que compone el gen CFTR producen una cantidad o función reducida de la proteína CFTR en la
superficie celular, pero el mecanismo específico es diferente entre las distintas clases de mutaciones, afectando los pasos del proceso de síntesis y trafico de
la proteína desde el núcleo a la membrana. Se han descripto mas de 2.000 mutaciones del gen, aunque no todas son causantes de FQ.
Una prueba de sudor patológica ó el hallazgo de 2 mutaciones del gen CFTR causantes de FQ en trans (una en cada alelo) junto a manifestaciones clínica o antecedentes familiares o pesquisa neonatal positiva confirma el diagnóstico.
La mutación F508del es la mas frecuente en los pacientes con FQ. Las mutaciones pueden estar presente en forma homocigota (la misma mutación
en los dos genes, ej F508del/F508del) o heterocigota (distintas mutaciones en cada uno de los dos genes, ej F508del/G542X)
Las mutaciones se agrupan funcionalmente en 6 clases de acuerdo con que paso de la síntesis de la proteína CFTR se afecta:
Clase 1: síntesis proteica truncada o ausente por una señal de terminación prematura (Ej G542X)
Clase 2: Alteración del procesamiento, con mal plegamiento de la proteína, maduración defectuosa, degradación prematura y con escasa llegada a la superficie celular (Ej F508del)
Clase 3: Alteración de la regulación de la proteína inserta en la membrana apical pero no activada por el AMPc y con tiempo de apertura reducido (Ej G551D)
Clase 4: Alteración de la conductividad y transporte iónico a través del canal (Ej R334W)
Clase 5: Expresión reducida de CFTR normal en la membrana (Ej R117H)
Clase 6: Recaptación acelerada del CFTR desde la membrana (Ej 1811+ 1.6 Kb A>G)
Si bien las mutaciones se la clasifican en un grupo funcional, muchas comparten características de más de una clase. Por ejemplo, F508del, que resulta en mínima expresión del canal en la membrana por alteración del procesamiento (clase 2), cuando llega a la membrana exhibe en su funcionamiento defecto de apertura (Clase 3) y una recaptación aumentada (Clase 6).
Hasta hace unos pocos años los tratamientos disponibles actuaban más allá de la membrana celular (mucolíticos como la Dornasa alfa, agentes osmóticos como el cloruro de sodio 7%, antibióticos inhalados como tobramicina o colistina, kinesiología respiratoria, etc.) en forma sintomática.
La cura de la enfermedad estaría dada por el reemplazo del gen mutado en el cromosoma (terapia génica), método no disponible hasta la actualidad. En los últimos años se han descubierto medicaciones (moduladores) que actúan modificando la función del canal CFTR.
Los moduladores son moléculas pequeñas que restauran parcialmente la función del canal CFTR mutado, evidenciado a través de ensayos in vitro o in vivo.
Existen dos tipos de moduladores: los potenciadores y los correctores. Los potenciadores mejoran la apertura del canal CFTR e incrementar el transporte del cloro cuando la proteína está inserta en la membrana apical. Los correctores actúan mejorando el tráfico intracelular de la proteína desde el retículo endoplásmico hacia la membrana celular.
Las terapias moduladoras del CFTR han sido desarrolladas para tratar la FQ en su defecto básico: el mal funcionamiento del canal. Ninguna de estas terapias son lo suficientemente efectivas como para usarlas como tratamientos únicos. Las terapias sintomáticas convencionales (antibióticos, mucolíticos,
kinesioterapia, soporte nutricional entre otras) que permiten a gran cantidad de pacientes llegar a la vida adulta, son de gran importancia.
Ivacaftor
Potenciador cuyo efecto es incrementar el tiempo en que el canal CFTR está abierto permitiendo así el transporte de cloro. Tiene eficacia en las mutaciones que producen defectos en la apertura. Inicialmente estudiada para la mutación G551D (clase 3), ha demostrado disminuir los valores de cloro en la prueba de sudor (prueba que actúa a nivel del canal), mejorar la función pulmonar (VEF1 y LCI), los puntajes de calidad de vida y el peso corporal y disminuir significativamente la ocurrencia de exacerbaciones respiratorias, que se sabe se asocian a una declinación progresiva y acelerada de la función pulmonar. En los estudios de extensión el efecto se mantuvo. Demostró efectividad en otras mutaciones asociadas a problemas de apertura.
Está indicada en pacientes con al menos una mutación que responda al ivacaftor basado en estudios clínicos o in vitro. Aprobado por la FDA a parir del año de vida y por ANMAT a partir de los 6 años.
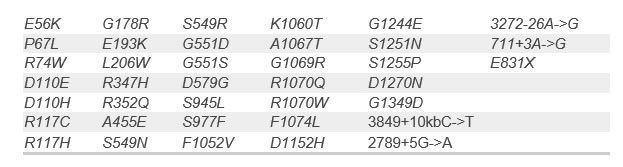
Listado de mutaciones respondedoras al ivacaftor
Dosis
Entre 2-6 años: 50 mg cada 12 hs (< 14 kg), 75 mg cada 12 hs (>14 kg) vía oral
Mayores de 6 años (aprobada en Argentina): 150 mg cada 12 hs vía oral
Debe ser ingerido con comidas grasas. Los eventos adversos más frecuentes fueron: cefalea, odinofagia, congestión nasal, dolor abdominal, diarrea, exantema, mareo, aumento de transaminasas. La administración continua es necesaria para ver los efectos de la medicación y su suspensión elimina los beneficios.
Lumacaftor/Ivacaftor
Es la asociación del potenciador ivacaftor y un corrector denominado lumacaftor, e incrementa el tiempo de apertura y mejora el tráfico del CFTR hacia la membrana apical respectivamente. Ha demostrado efectividad en pacientes homocigotas para la mutación F508del, mejorando la función pulmonar medida a través del VEF1 o LCI, reduciendo significativamente las exacerbaciones y con una tasa menor de declinación del VEF1 comparado con controles.
Ha sido aprobado por la FDA a partir de los 2 años y por ANMAT a partir de los 12 años.
Se indica en pacientes con dos copias de la mutación F508del.
Dosis:
6 a 11 años: 400 mg de lumacaftor + 500 mg de ivafactor cada 12 hs
Mayores de 12 años (autorizado en Argentina): 800 mg de lumacaftor + 500 mg de ivafactor cada 12 hs
También debe ser ingerido con comidas grasas. Los efectos adversos más comunes fueron: tos, disnea, nasofaringitis, nausea, diarrea, opresión torácica; esta última es más frecuente pacientes con gran afectación respiratoria ó en las primeras semanas de tratamiento pudiéndose disminuir la dosis para después incrementarla gradualmente ó comenzar a mitad de la dosis.
Reducir la dosis en afectación hepática moderada o grave. Se deben realizar controles de función hepática, tensión arterial, fondo de ojo.
Tezacaftor +Ivacaftor
Es la asociación de un corrector (tezacaftor) y un potenciador (ivacaftor).
Indicado en pacientes homocigotas F508de (especialmente con afectación hepática) ó pacientes con al menos una de las 26 mutaciones que responda al tezacaftor+ ivacaftor basado en datos clínicos e in vitro. Produce mejoría de la función pulmonar, calidad de vida y reducción de 35% de las exacerbaciones respiratorias.
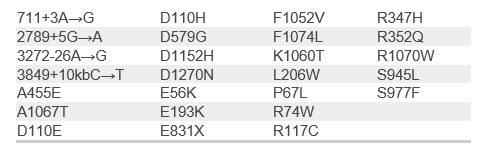
Listado de mutaciones respondedoras a Tezafactor + Ivacaftor
Dosis:
100 mg de tezafactor+ 150 mg de ivacaftor a la mañana y 150 mg de ivacaftor a la noche
(dosis diaria: Tezafactor 100 mg + Ivacaftor 300 mg)
Entre los efectos adversos se reportaron cefalea, congestión sinusal y mareos. Produce menor sensación de opresión torácica comparado con el lumacaftor + ivacaftor.
Todavía no está aprobado en la Argentina
Nuevas asociaciones
Actualmente se encuentran en estudio de fase 3, triple combinaciones de 2 correctores (tezacaftor y VX-659) y un potenciador (ivacaftor) en pacientes con una copia de F508del y una mutación de función mínima que en los resultados preliminares han demostrado un aumento del VEF1 de 14 puntos porcentuales luego de 4 semanas de tratamiento y en pacientes con dos copias de F508 del un aumento de 10 puntos comparado con el grupo que solo recibió tezacaftor + ivacaftor.